QSAR structure-property model
Predicting the inhibitory activity of molecules
artificial intelligence
ABOUT PRODUCT
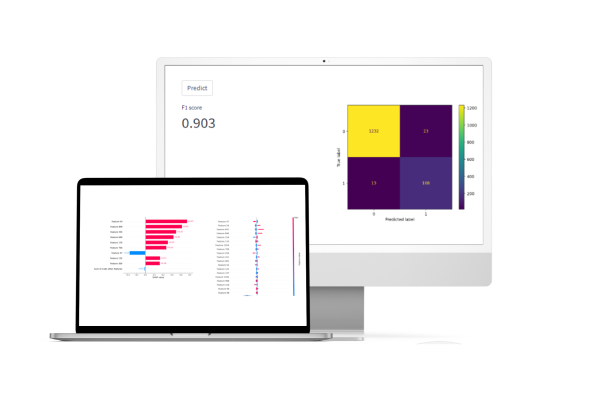
Software that predicts the inhibitory activity of molecules using QSAR models. Half-inhibition concentration, binary classification is identified.
INDUSTRIES
PHARMACEUTICAL INDUSTRY
Research time: 1 target - 1 month
Gradient bousting of symmetric solving trees is used.
Result: list with molecules and selected inhibitors + IP analysis and ADMET.
Result: list with molecules and selected inhibitors + IP analysis and ADMET.
Addresses


Accelerates preclinical development of medication.
Reduces inhibitor selection time per 1 molecule from 10 seconds to 0.01 seconds

5-6x less
reduces time and resources to obtain inhibitor list
increases preclinical studies accuracy
>30%
Effects
Product benefits
Proprietary architecture
Ability to work with a small dataset
High prediction accuracy



CASES
Building a QSAR model for the biological target MCL-1 for ChemRar
2.12.2022 / media.innopolis.university
FAQ
QSAR is a method that is used to predict the biological activity of molecules based on their structural properties.
The customer provides the following information: the biomimic of interest, its properties, information on whether IP validation is needed.
ADMET filtering, evaluation by MCE-18, RO5, novelty, SA/ReRSA scores and T-indexes.
The customer receives a document with sorted molecules for one target, where the following are presented: activity of molecules, toxicity of molecules, patent purity testing, retrosynthesis possibilities.
With docking, approximately 10 seconds are spent per molecule, the QSAR model reduces this time to 0.01 seconds.
QSAR is a method that is used to predict the biological activity of molecules based on their structural properties.
The customer provides the following information: the biomimic of interest, its properties, information on whether IP validation is needed.
ADMET filtering, evaluation by MCE-18, RO5, novelty, SA/ReRSA scores and T-indexes.
The customer receives a document with sorted molecules for one target, where the following are presented: activity of molecules, toxicity of molecules, patent purity testing, retrosynthesis possibilities.
With docking, approximately 10 seconds are spent per molecule, the QSAR model reduces this time to 0.01 seconds.
Submit a product request

Email
Telephone
Newsletter for those who want to know
everything and a little more about technology

